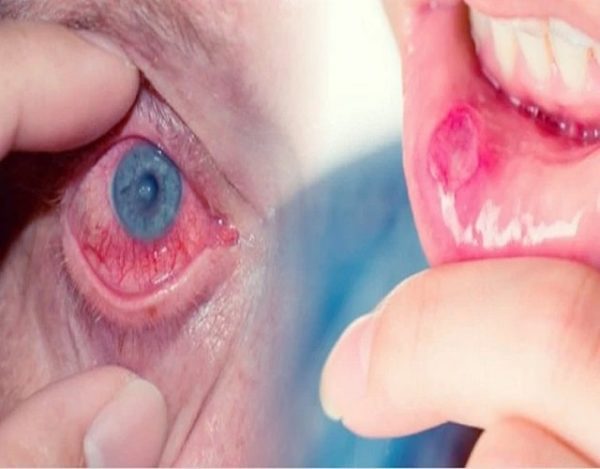
En la década de 1930, un dermatólogo turco, Hulusi Behcet, observó la tríada de úlceras orales aftosas, lesiones genitales e inflamación recurrente de los ojos, y se convirtió en el primer médico en describir la enfermedad de Behcet en los tiempos modernos.
Este síndrome es una de las pocas formas de vasculitis en la que existe una predisposición genética conocida. La presencia del gen HLA-B51 es un factor de riesgo para esta enfermedad. Sin embargo, debe enfatizarse que la presencia del gen en sí misma no es suficiente para causar el Behcet: muchas personas poseen el gen, pero pocos desarrollan la enfermedad. Se cree que otros factores (quizás más de uno) juegan un papel importante. Las posibilidades incluyen infecciones y otras exposiciones ambientales.
Índice
¿Quién contrae la enfermedad de Behcet (el paciente «típico»)?
La enfermedad de Behcet es más común a lo largo de la «Ruta de la Seda Vieja», que abarca la región desde Japón y China en el Lejano Oriente hasta el Mar Mediterráneo, incluidos países como Turquía e Irán. Esta afección es muy rara en Estados Unidos, esporádicamente solo se reportan casos de pacientes que no tienen ningún factor de riesgo. La enfermedad no es rara en las regiones a lo largo de la Ruta de la Seda Antigua, pero la epidemiología de la enfermedad no se conoce bien. En Japón, la enfermedad de Behcet es una de las principales causas de ceguera.
Síntomas y signos clásicos de la enfermedad
La enfermedad de Behcet es prácticamente incomparable entre las vasculitis en su capacidad de involucrar vasos sanguíneos de casi todos los tamaños y tipos, que van desde arterias pequeñas a grandes, y que también involucran venas. Las manifestaciones de Behcet pueden ocurrir en muchos sitios en todo el cuerpo, hay predilección por ciertos órganos y tejidos:
Ojos
Puede causar uveítis anterior (inflamación en la parte frontal del ojo) o uveítis posterior (inflamación en la parte posterior del ojo), y a veces causa ambas cosas al mismo tiempo. La uveítis posterior puede ser más peligrosa y poner en riesgo la visión porque a menudo causa menos síntomas y daña una parte crucial del ojo: la retina.
Boca
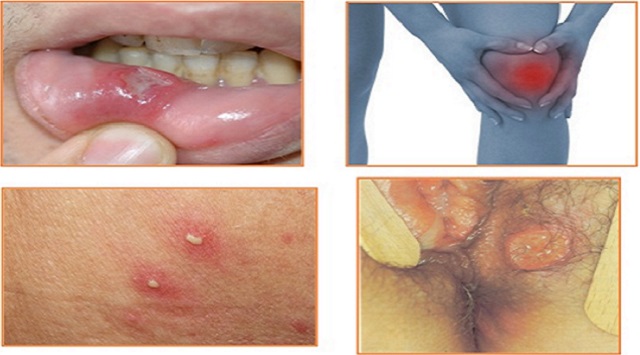
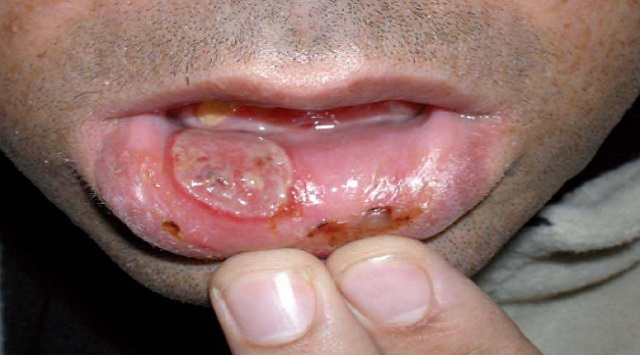
Llagas dolorosas en la boca llamadas úlceras aftosas. Las lesiones son más numerosas, más frecuentes, y a menudo más grandes y más dolorosas.
Piel
Lesiones cutáneas pustulares que se asemejan al acné, pueden aparecer en casi cualquier parte del cuerpo. Las lesiones de la enfermedad de Behcet con frecuencia se ulceran.
Articulaciones
Artritis o «artralgias» (dolor en las articulaciones no acompañado de hinchazón articular).
Cerebro
La afectación del sistema nervioso central es una de las manifestaciones más peligrosas. Tiende a involucrar la porción de materia blanca del cerebro y el tronco encefálico, y puede provocar dolores de cabeza, confusión, derrames cerebrales, cambios de personalidad y (rara vez) demencia.
Genitales
- Masculino: lesiones genitales dolorosas que se forman en el escroto, similar a las lesiones orales, pero más profundas.
- Femenino: úlceras genitales dolorosas que se desarrollan en la vulva.
Tracto gastrointestinal
Las ulceraciones pueden ocurrir en cualquier parte del tracto gastrointestinal desde la boca hasta el ano. El íleon terminal y el ciego son sitios comunes. La participación del tracto gastrointestinal por la enfermedad de Behcet puede ser difícil de distinguir de la enfermedad inflamatoria intestinal (como la enfermedad de Crohn).
¿Cómo se diagnostica la enfermedad de Behcet?
No hay una prueba específica para diagnosticar las de Behcet. Más bien, el diagnóstico se basa en la aparición de síntomas y signos que son compatibles con la enfermedad. La presencia de ciertas características que son particularmente características (p. Ej., Ulceraciones orales o genitales), la eliminación de otras posibles causas del síntoma del paciente y, si es posible, la prueba de vasculitis por biopsia de un órgano involucrado, respaldarían un diagnóstico de Behcet.
Tratamiento y curso de la enfermedad de Behcet

Para la enfermedad que se limita a las regiones mucocutáneas (boca, genitales y piel), los esteroides tópicos y los medicamentos no inmunosupresores como la colchicina pueden ser efectivos. También se requieren dosis moderadas de corticosteroides sistémicos para las exacerbaciones de la enfermedad, y algunos pacientes requieren dosis bajas y crónicas de prednisona para mantener la enfermedad bajo control.